Bei Wang, Arabella H. Wan, Yu Xu, Ruo-Xin Zhang, Ben-Chi Zhao, Xin-Yuan Zhao, Yan-Chuan Shi, Xiaolei Zhang, Yongbo Xue, Yong Luo, Yinyue Deng, G. Gregory Neely, Guohui Wan & Qiao-Ping Wang
Abstrakt
Der „Todespilz“, Amanita phalloides, ist der giftigste Pilz der Welt und für 90 % der durch Pilze verursachten Todesfälle verantwortlich. Der tödlichste Bestandteil des Knollenblätterpilzes ist α-Amanitin. Trotz seiner tödlichen Wirkung bleiben die genauen Mechanismen, wie α-Amanitin den Menschen vergiftet, unklar, so daß kein spezifisches Gegenmittel für die Behandlung verfügbar ist. Hier wird gezeigr, daß STT3B für die α-Amanitin-Toxizität erforderlich ist und sein Inhibitor Indocyaningrün (ICG) als spezifisches Gegenmittel verwendet werden kann. Durch die Kombination eines genomweiten CRISPR-Screenings mit einem In-silico-Wirkstoffscreening und einer In-vivo-Funktionsvalidierung entdeckte das Forscherteam, daß der N-Glykan-Biosyntheseweg und seine Schlüsselkomponente STT3B eine entscheidende Rolle bei der α-Amanitin-Toxizität spielen und dass ICG ein STT3B Inhibitor ist.
Darüber hinaus zeigten die Ergebisse, daß ICG die toxische Wirkung von α-Amanitin in Zellen, Leberorganoiden und männlichen Mäusen wirksam blockiert, was zu einer Gesamtüberlebenssteigerung der Tiere führt. Durch die Kombination eines genomweiten CRISPR-Screenings auf α-Amanitin-Toxizität mit einem In-silico-Arzneimittelscreening und einer funktionellen Validierung in vivo hebt die Studie ICG als STT3B-Inhibitor gegen das Pilztoxin hervor.
Einführung
Pilzvergiftungen sind weltweit die Haupttodesursache bei Lebensmittelvergiftungen. Zwischen 2010 und 2020 wurden in China insgesamt 10.036 Expositionsereignisse gemeldet, die zu 38.676 Erkrankungen und 788 Todesfällen führten. Unter allen giftigen Pilzen sind Knollenblätterpilze (Amanita phalloides) für mehr als 90 % aller Todesfälle verantwortlich. Eine Amatoxinvergiftung ist häufig mit schlechten Ausgang verbunden, hauptsächlich aufgrund des irreparablen akuten Versagens der Leber oder der Niere.
α-Amanitin (AMA) ist eines der giftigsten Amatoxine. Es wird angenommen, daß die toxischen Wirkungen von AMA auf den Menschen mit der Hemmung der RNA-Polymerase II (RNAP II) verbunden sind, was zur Produktion von Tumornekrosefaktor-α (TNFα)8, oxidativem Stress9 und Apoptose führt. Traditionelle Therapien beschränken sich oft auf die unspezifische Neutralisierung von Toxinen sowie symptomatische und unterstützende Pflege.
In den letzten Jahrzehnten haben mehrere klinische Medikamente, darunter Silybin und Penicillin, eine starke therapeutische Wirksamkeit bei menschlichen Amatoxinvergiftungen gezeigt, obwohl die genauen Wirkmechanismen unklar bleiben. Darüber hinaus wurde gezeigt, daß Polymyxin B, das in einem virtuellen Docking als potenzieller RNAP-II-Inhibitor identifiziert wurde, die AMA-Toxizität bei Mäusen blockiert. Spezifische Gegenmittel, die auf bestimmte Proteine abzielen, die eine entscheidende Rolle bei der AMA-Toxizität spielen, sind jedoch nicht verfügbar, da ein vollständiges molekulares Verständnis der AMA-Zytotoxizität fehlt.
Kürzlich haben CRISPR-Screenings (Pooled Clustered Regular Interspaced Short Palindromic Repeats) das molekulare Verständnis der molekularen Mechanismen, die den Zelltod steuern, beschleunigt. Diese Hochdurchsatz-CRISPR-Screenings wurden häufig zur Identifizierung von Genen oder Signalwegen eingesetzt, die an Arzneimittelresistenzen, Mechanismen von Bakterientoxinen oder Virusinfektionen beteiligt sind. Darüber hinaus haben die Wissenschaftler diesen Ansatz genutzt, um die molekularen Mechanismen des tödlichen Quallengifts zu analysieren, was zu einem wirksamen Gegenmittel gegen die Quallentoxizität führte.
In dieser Studie soll ein systematischenr Rahmen für die Entwicklung von Gegenmitteln geschaffen werden, indem die Identifizierung neuer Wirkstoffziele mithilfe eines genomweiten CRISPR-Screenings und eines virtuellen Screenings von FDA-zugelassenen Medikamenten kombiniert wird. Die Wissenschaftler führten zunächst ein genomweites CRISPR-Funktionsverlust-Screening durch, um Gene und Signalwege zu identifizieren, die an der AMA-Zytotoxizität beteiligt sind. Sie fanden heraus, daß mehrere neuartige Wege, einschließlich der N-Glykan-Biosynthese und des Cholesterinstoffwechsels, am AMA-induzierten Zelltod beteiligt sind. Es wurde außerdem dargestellt, daß die N-Glykan-Biosynthese und sein katalytisches Enzym STT3B für die AMA-Toxizität erforderlich sind. Durch die Kombination dieser Daten mit einem In-silico-Screening von FDA-zugelassenen Arzneimitteln und einer anschließenden Funktionsvalidierung konnten die Forscher ICG erfolgreich als potenziellen STT3B-Inhibitor identifizieren. Sie haben außerdem gezeigt, daß ICG den AMA-induzierten Zelltod in vivo und in vitro blockieren kann, was darauf hindeutet, daß ICG bei der Behandlung von AMA/Todeskappenvergiftungen nützlich sein könnte.
Ergebnisse
Ein Hochdurchsatz-CRISPR-Screening zur Identifizierung von Genen und Signalwegen, die für den AMA-induzierten Zelltod erforderlich sind
Amanita phalloides ist eine häufige Todesursache bei Lebensmittelvergiftungen, vor allem aufgrund der Produktion von AMA26 (Abb. 1a). Um die Schlüsselgene und -wege zu identifizieren, die für den AMA-induzierten Zelltod erforderlich sind, führte das Forscherteam unter Verwendung der gepoolten menschlichen CRISPR-Knockout-Bibliothek (Brunello) ein genomweites Funktionsverlust-Screening durch, das auf insgesamt 19.114 Gene abzielt. Sie haben ihr Screening mit der haploiden Zelllinie HAP1 durchgeführt, die ausgiebig zur Untersuchung der Mechanismen der Arzneimittelresistenz, der Toxikologie, der synthetischen Letalität und der Virusinfektion eingesetzt wurde. Vor dem Screening haben sie zunächst die 50-prozentige Hemmkonzentration (IC50) von AMA bestimmt HAP1-Zellen (Abb. 1b).
Anschließend haben sie HAP1-Zellen mit der Brunello-Bibliothek bei einer niedrigen Infektionsmultiplizität (MOI ≈ 0,3) transduziert, um sicherzustellen, dass die meisten HAP1-Zellen nur eine genetische Störung erfahren (Abb. 1c). Mittlerweile wurde die Abdeckung von >500 Zellen sichergestellt, die jeweils 77.441 sgRNA exprimieren. Transfizierte Zellen wurden dann 7 Tage lang mit 1 μg/ml Puromycin selektiert. Anschließend wurden mutierte Zellpools 7 Tage lang einer Dosis von 1,5 μM AMA ausgesetzt und die genomische DNA wurde aus den überlebenden Zellen zur Tiefensequenzierung extrahiert.
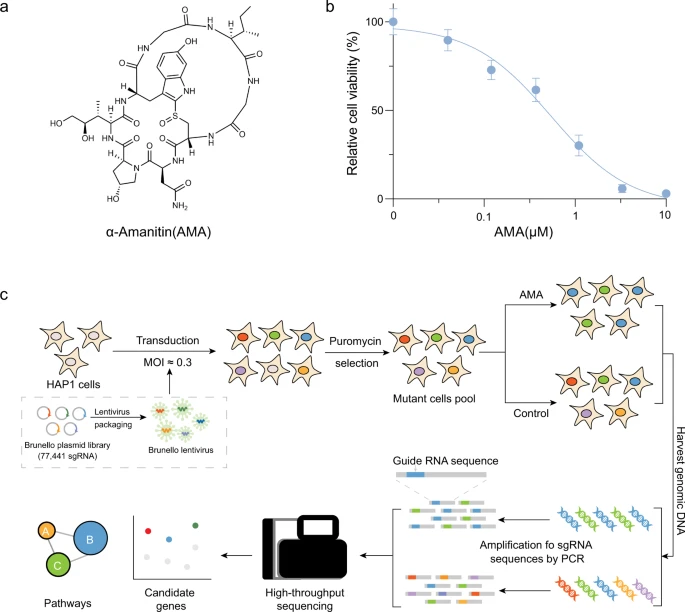
Nach einer modellbasierten Analyse der genomweiten CRISPR/Cas9-Knockout-Analyse (MAGeCK) haben sie Hunderte von Genen identifiziert, die mit dem AMA-induzierten Zelltod assoziiert sind (Abb. 2a, b, Ergänzende Daten 1). Eine Untergruppe von sgRNAs, die auf 559 Gene abzielen, war in den mit AMA behandelten Zellen im Vergleich zu den unbehandelten Kontrollen signifikant verändert (p < 0,05 und |LFC | > 1), was darauf hinweist, daß diese Gene an der AMA-Toxizität beteiligt waren (Abb. 2c).
Die Forscher haben zunächst die bioinformatischen Analysen der Genontologie (GO) und KEGG an diesen veränderten Genen durchgeführt. Wie erwartet war „Regulation der Transkription vom RNA-Polymerase-II-Promotor“ am stärksten im biologischen Prozess angereichert (Abb. 2d), und „RNA-Polymerase-II-Transkriptionsfaktor-Aktivitätssequenzspezifische DNA-Bindung“ war auch am stärksten in der molekularen Funktion angereichert Klassifikator (Abb. 2e). Dies steht im Einklang mit dem bisherigen Verständnis, daß AMA durch Hemmung der RNA-Polymerase II31 wirkt. Mehrere KEGG-Wege, einschließlich Apoptose, N-Glykan-Biosynthese und Cholesterinstoffwechsel, wurden ebenfalls durch AMA-induzierten Zelltod angereichert (Abb. 2f).
Das Forscherteam verwendete außerdem einen Netzwerkausbreitungsansatz, um das Gennetzwerk zwischen diesen angereicherten KEGG-Pfaden besser zu verstehen (Abb. 2g). Unter diesen angereicherten Prozessen oder Signalwegen wurden die Transkription und Apoptose der RNA-Polymerase II und die Apoptose10,32 mit der AMA-Toxizität in Verbindung gebracht, über die N-Glykan-Biosynthese und den Cholesterinstoffwechsel wurde jedoch noch nicht berichtet, was darauf hindeutet, daß diese beiden Signalwege eine entscheidende Rolle bei der AMA-Toxizität spielen könnten. Gemeinsam hat das Screening neue Wege identifiziert, die für den AMA-induzierten Zelltod erforderlich sind.
Für den AMA-induzierten Zelltod ist die N-Glykan-Biosynthese erforderlich
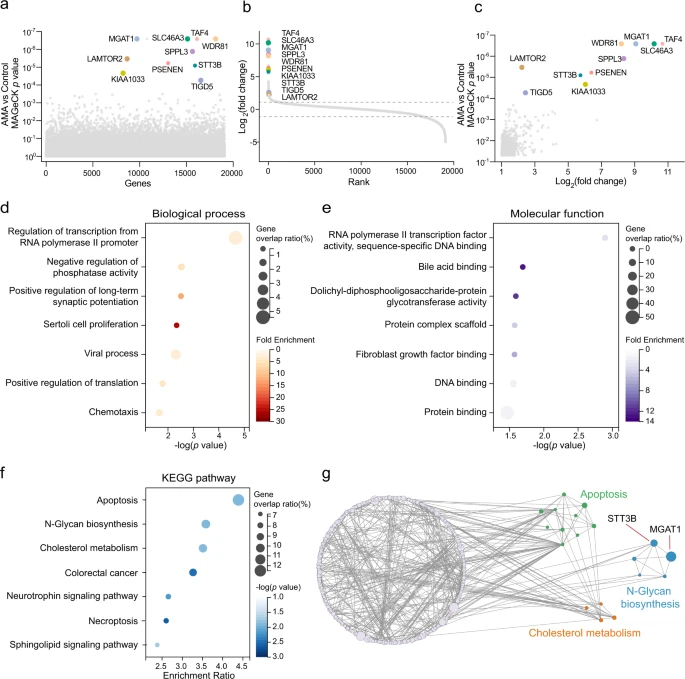
Der N-Glykan-Biosyntheseweg ist von besonderem Interesse, da seine Schlüsselkomponenten STT3B und MGAT1 in den Top-10-Genen angereichert waren. N-Glykane auf Glykoproteinen dienen als eine der wichtigsten ko- und posttranslationalen Proteinmodifikationen in eukaryotischen Zellen33 und haben mehrere biologische Funktionen, wie Zelladhäsion, intrazelluläre Signalübertragung, Homöostase und Entzündung34,35. Die N-Glykan-Biosynthese findet im endoplasmatischen Retikulum (ER) und im Golgi-Apparat statt und erfordert eine Reihe von Prozessen, die durch mehrere Enzyme katalysiert werden, darunter ALG10, STT3A/3B, MAN1, MGAT1, MAN2 und MGAT2 (Abb. 3a)36. Wir fanden heraus, daß die Zählungen für sgRNA, die auf ALG10, STT3A/3B, RPN2 und MGAT1 abzielen, in AMA-behandelten Zellen angereichert waren (ergänzende Abbildung 1a – e) und dass STT3B und MGAT1 in den Top-10-Treffern angereichert waren.
Um die Rolle der N-Glykan-Biosynthese beim AMA-induzierten Zelltod zu validieren, verwendeten die Wissenschaftler zunächst Kifunensin (KIF), einen wirksamen Inhibitor kleiner Alkaloide, um die MAN1-Aktivität pharmakologisch zu hemmen und so die Synthese von N-Glykanen zu blockieren37,38. Wie erwartet zeigten KIF-behandelte Zellen eine Resistenz gegen den AMA-induzierten Tod in HAP1-Zellen (Abb. 3b). Wichtig ist, dass KIF auch den Zelltod in HAP1-Zellen hemmte, die 6 Stunden lang mit AMA vorbehandelt wurden (Abb. 3c).
STT3B ist eine vorgelagerte Komponente des N-Glykan-Biosynthesewegs39. STT3B und STT3A bilden den Oligosaccharyltransferase (OST)-Komplex, der die posttranslationale Glykosylierung in ER40,41 katalysiert. Um die Rolle von STT3B bei der AMA-Toxizität weiter zu bestätigen, haben wir mithilfe der CRISPR-Cas9-Technologie STT3B-Knockout-HAP1- und HepG2-Zelllinien generiert. Dementsprechend war die STT3B-mRNA-Expression in beiden Zellen stark reduziert (ergänzende Abbildung 2a, b). Die Abreicherung von STT3B führte zu einer erhöhten Resistenz gegen AMA-induzierten Zelltod in HAP1-Zellen (Abb. 3d) und HepG2-Zellen (Abb. 3e).
Das Forscherteam hat diese Ergebnisse in HepG2-Zellen mit STT3B-Knockdown unter Verwendung von Short-Hairpin-RNA (shRNA) weiter bestätigt (ergänzende Abbildung 2c). Da STT3B bei der Proteinglykosylierung mit STT3A zusammenarbeitet, fragten sie sich, ob die Abreicherung von STT3A in STT3B-Knockout-Zellen zu einer stärkeren Resistenz gegen AMA-Zytotoxizität führen könnte. Als STT3A in STT3B-Knockout-HepG2-Zellen ausgeschaltet wurde, erlangten diese Zellen eine nahezu vollständige Resistenz gegen den AMA-induzierten Zelltod (ergänzende Abbildung 2d-e), was darauf hindeutet, dass die teilweise Resistenz von STT3B-Knockout-HepG2 gegen AMA auf die Expression von STT3A zurückzuführen war OST-Komplex, der funktionell redundant zum STT3B OST-Komplex40 ist.
Darüber hinaus verwendeten sie einen niedermolekularen N-verknüpften Glykosylierungsinhibitor 1 (NGI-1), um die Aktivität von OST42,43 zu blockieren. NGI-1 kann den AMA-induzierten Zelltod bei 2,5 μM hemmen. NGI-1 selbst war jedoch bei 5–20 μM toxisch für Zellen (ergänzende Abbildung 2f), was darauf hindeutet, dass NGI-1 nicht als Gegenmittel zur AMA-Toxizität verwendet werden konnte . Als nächstes fragten sie sich, ob die Glykanbiosynthese den Eintritt von AMA in Zellen beeinflusst. Der intrazelluläre AMA-Gehalt wurde mit einem etablierten Hochleistungsflüssigkeitschromatographietest (HPLC) quantifiziert (ergänzende Abbildung 3). Die Abreicherung von STT3B verringerte den Eintritt von AMA in HAP1-Zellen (Abb. 3f) und HepG2-Zellen (Abb. 3g) signifikant.
Unterdessen hat STT3B-Knockout keinen Einfluss auf die Expression von OATP1B3 und NTCP, die AMA-Transporter in diesen Zellen sind (Abb. 3h, i). Die Hemmung der Glykosylierung kann Stressreaktionen im Endoplasmatischen Reticulum und im Golgi-Apparat auslösen 44, 45, 46. Der STT3B-Knockout löste jedoch weder im ER noch im Golgi Stressreaktionen aus (ergänzende Abbildung 4). Zusammengenommen deuten alle Daten darauf hin, daß STT3B für den AMA-induzierten Zelltod erforderlich ist und den Eintritt von AMA in Zellen beeinflusst. In-silico-Screening von FDA-zugelassenen Molekülen für den STT3B-Inhibitor: Da die Blockierung der N-Glykan-Biosynthese den AMA-induzierten Zelltod verhindern kann, wären die STT3B-Inhibitoren potenzielle Gegenmittel zur Behandlung der AMA-Toxizität. Bisher wurde über kein von der FDA zugelassenes Molekül berichtet, das STT3B spezifisch hemmt. Daher führten die Wissenschaftlerr ein In-silico-Screening von von der FDA zugelassenen Molekülen durch, um nach potenziellen STT3B-Inhibitoren zu suchen.
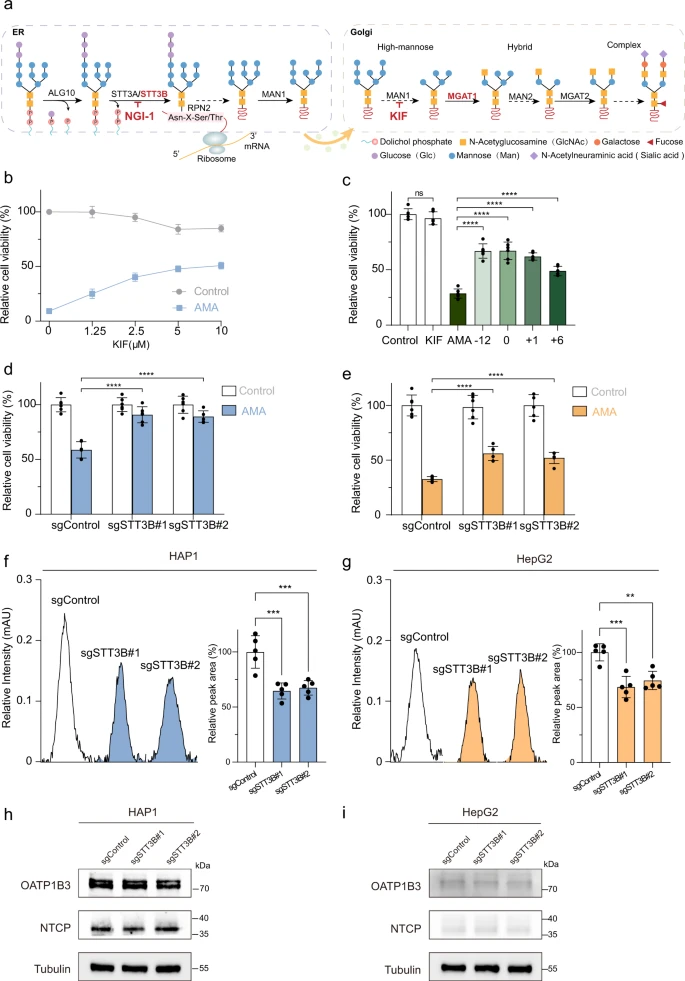
Für das virtuelle Screening auf STT3B-Inhibitoren wurden die FDA-Molekülbibliotheken (ZINC und Drugbank) mit insgesamt 3201 Verbindungen verwendet. In STT3B gab es zwei mutmaßliche Bindungstaschen. Nach dem In-silico-Screening wurden insgesamt die 34 besten Verbindungen für die zelluläre In-vitro-Validierung ausgewählt (Abb. 4a, Zusatzdaten 2). Aufgrund der Nichtverfügbarkeit einiger Verbindungen wurden nur 24 Verbindungen auf ihren zellulären Schutz gegen AMA-Toxizität getestet. Von allen getesteten Medikamenten konnten ICG und Posaconazol den Zelltod in HAP1-Zellen signifikant verhindern (Abb. 4b), ohne dass die Zellen zusätzlich zytotoxisch wirkten (Abb. 4c). ICG bot nahezu vollständigen Schutz gegen die AMA-Zytotoxizität.
Beim molekularen Andocken bindet ICG an STT3B und blockiert möglicherweise die katalytische Aktivität. ICG hat mehrere Kontakte mit STT3B (Abb. 4d, e), einschließlich der drei Sauerstoffatome von zwei Sulfobutyleinheiten, die drei Wasserstoffbrückenbindungen mit Seitenketten von Ser319, Trp380 und Ser449 gebildet haben. Darüber hinaus beteiligte sich der Benzolring der Benzoindolyleinheit in ICG an einem versetzten Face-to-Face-Pi-Stapel mit der Seitenkette von Trp380 in STT3B. Die hemmende Wirkung von ICG wird durch die Besetzung des Eingangs der STT3B-Substratbindungstasche vermittelt.
ICG lindert die AMA-Toxizität in Zellen und Leberorganoiden
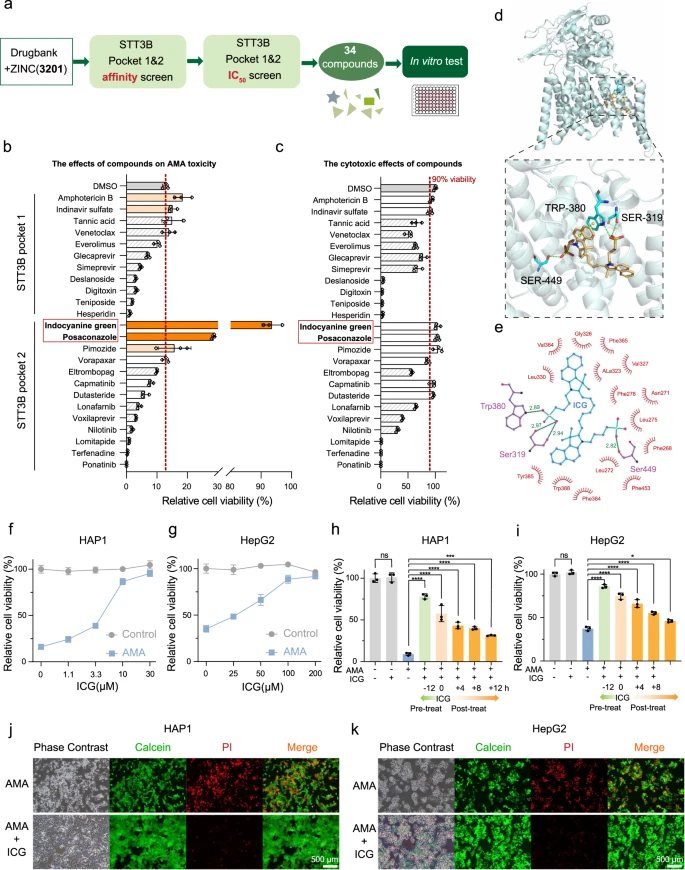
ICG, ein fluoreszierender Jodidfarbstoff, ist seit 1956 von der FDA als diagnostisches Reagenz beim Menschen zugelassen und wird heute häufig in der Augenangiographie und der Beurteilung der Leberfunktion eingesetzt47. ICG kann schnell von Hepatozyten abgebaut werden47 und ICG hat bei einer Standard-Einzeldosis von 0,5 mg/kg keine offensichtlichen Nebenwirkungen (50 % tödliche Dosis beträgt 60-80 mg/kg für Mäuse)48. Daher haben die Forscher die therapeutische Wirkung von ICG auf die AMA-Toxizität weiter untersucht.
Die ICG-Vorbehandlung führte zu einer dosisabhängigen Verringerung des Zelltods sowohl in HAP1-Zellen (Abb. 4f) als auch in HepG2-Zellen (Abb. 4g). Darüber hinaus hemmten ICG-Behandlungen auch den Zelltod in AMA-vorbehandelten HAP1-Zellen (Abb. 4h) und HepG2-Zellen (Abb. 4i) signifikant. Um weiter zu bestätigen, daß ICG die AMA-Toxizität blockieren kann, überwachte das Forscherteam den Zelltod mithilfe der Calcein/Propidiumiodid (PI)-Färbung und stellte erneut fest, daß mit ICG vorbehandelte Zellen viel resistenter gegen den AMA-induzierten Zelltod waren (Abb. 4j, k).
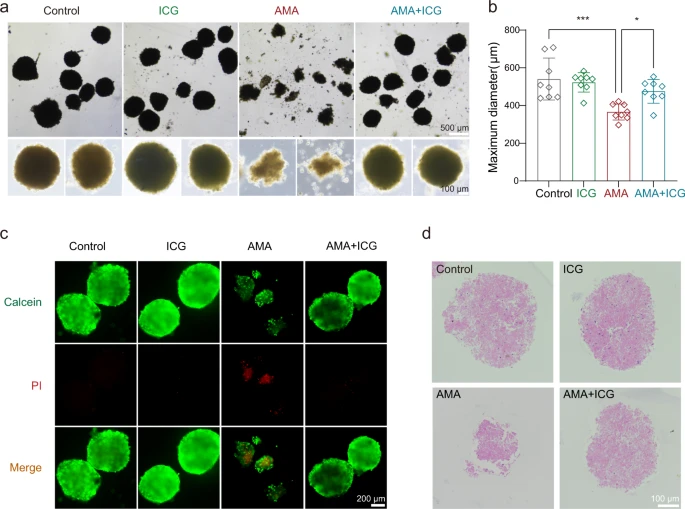
Darüber hinaus haben sie ein Mausleber-Organoidmodell zur weiteren Bewertung der therapeutischen Wirkung von ICG erstellt. In Übereinstimmung mit ihren Beobachtungen in HAP1- und HepG2-Zellen könnte ICG die zytotoxische Wirkung von AMA auf diese Leberorganoide wirksam blockieren.
Mit ICG behandelte Organoide waren enger verbunden und größer als die mit Vehikel behandelten Organoide (Abb. 5a, b). Wir beobachteten auch, dass die ICG-Behandlung den AMA-induzierten Zelltod durch den Calcein/PI-Färbetest signifikant verhinderte (Abb. 5c). Der ICG-Behandlungseffekt wurde auch durch Hämatoxylin- und Eosin-Färbung (H&E) beobachtet und ICG blockierte die AMA-Toxizität auf Organoiden (Abb. 5d). Zusammengenommen ist die Kombination des funktionellen CRISPR-Screenings mit der In-silico-Arzneimittelvorhersage eine praktikable Pipeline zur schnellen Identifizierung neuer Toxin-Gegenmittel. Hier zeigen die Forscher, daß ICG ein potenzieller STT3B-Inhibitor ist, der den AMA-induzierten Zelltod verhindern kann.
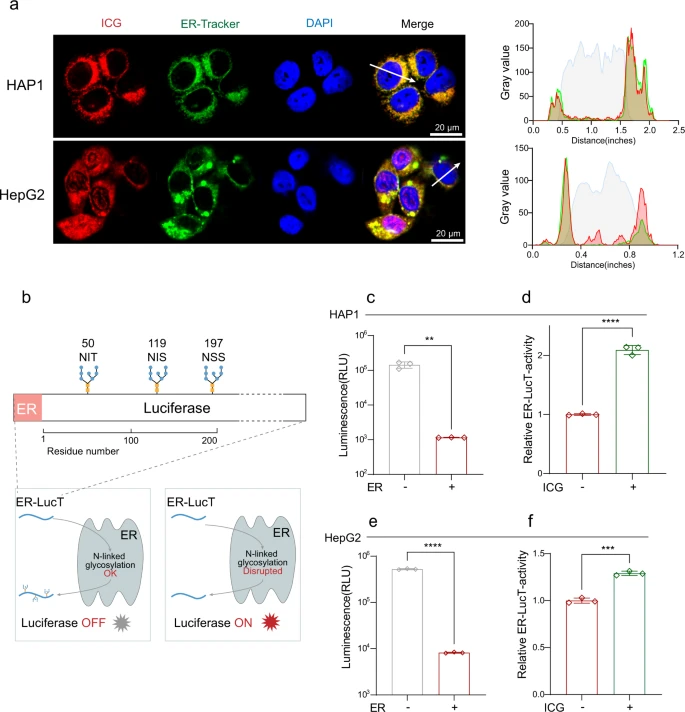
ICG verhindert den AMA-induzierten Zelltod, indem es die STT3B-Aktivität hemmt Um zu testen, ob ICG mit STT3B in Zellen interagieren kann, wurde die intrazelluläre Lokalisierung von ICG nachgewiesen. ICG kann grüne Fluoreszenz erzeugen. Es wurde heraus gefunden, daß ICG in HAP1-Zellen zusammen mit ER lokalisiert war, und diese Kolokalisation wurde in HepG2-Zellen bestätigt (Abb. 6a), was darauf hindeutet, daß ICG im ER wirkt, wo auch STT3B lokalisiert ist.
Um weiter zu testen, ob ICG die STT3B-Aktivität hemmen kann, verwendete das Wissenschaftsteam einen Biolumineszenz-Reporter, nämlich ER-LucT, um die STT3B-Aktivität durch ein ER-LucT-Reportersystem zu bewerten42,43. Dieses System besteht aus einer modifizierten Luciferase (Luc) mit einer ER-Translationssequenz und drei (T) potenziellen Glykosylierungsstellen49. Die N-Glykosylierung der modifizierten Luciferase hemmt deren Aktivität und reduziert die Biolumineszenz (Abb. 6b).
Das ER-LucT-Reportersystem wurde zunächst in HAP1-Zellen getestet. Die Lumineszenz war in Zellen mit ER-LucT signifikant verringert, was darauf hindeutet, daß die Luciferase-Aktivität gehemmt war (Abb. 6c). Wie erwartet erhöhte ICG im Einklang mit NGI-1 (ergänzende Abbildung 5a) die Aktivität von ER-LucT, indem es die durch STT3B vermittelte N-Glykosylierung verringerte (Abb. 6d). Ähnliche Ergebnisse wurden auch in HepG2-Zellen beobachtet (Abb. 6e, f, ergänzende Abb. 5b). Zusammen verhindert ICG den AMA-induzierten Zelltod der Zellen, indem es die STT3B-Aktivität hemmt.
ICG ist ein wirksames Gegenmittel zur Behandlung der AMA-Toxizität bei Mäusen Als nächstes testeten die Forscher die Wirksamkeit von ICG als AMA-Gegenmittel an Tieren. Um die tatsächliche AMA-Toxizität beim Menschen nachzuahmen50,51, wurde ICG Mäusen verabreicht, die wie zuvor berichtet 4 Stunden lang mit intraperitonealem (i.p.) AMA mit 0,33 mg/kg vorbehandelt wurden5,52.
Drei aufeinanderfolgende Verabreichungen von ICG in einer Menge von 5 mg/kg, was etwa 0,5 mg/kg beim Menschen entspricht53, wurden Mäusen im Abstand von 4 Stunden intravenös (iv) injiziert (Abb. 7a). Die ICG-Verteilung wurde durch Nahinfrarot (NIR)54 überwacht. Nach der Injektion verteilte sich ICG schnell im ganzen Körper und konzentrierte sich nach 2 Stunden hauptsächlich in der Leber (Abb. 7b), was mit früheren Beobachtungen übereinstimmt, daß ICG schnell aus dem Plasma entfernt und selektiv von der Leber aufgenommen wird55.
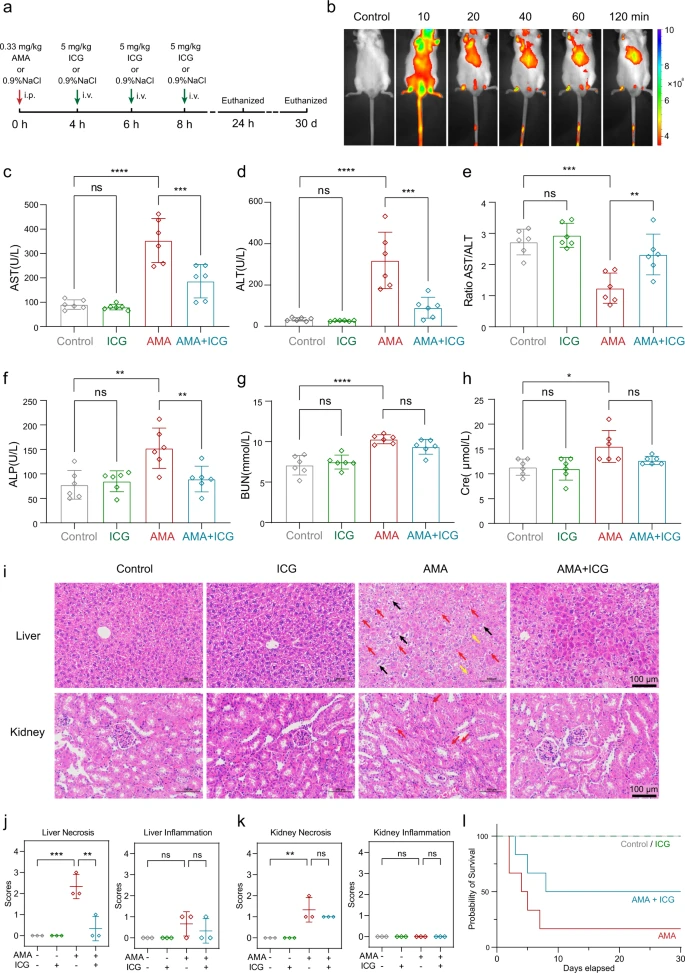
Da Leber und Niere die wichtigsten AMA-Zielorgane sind, wurde die schützende Wirkung von ICG auf die AMA-exponierte Leber und Niere untersucht. Die Schäden in Leber und Niere wurden durch Messung von Plasma-Biomarkern und histopathologischen Analysen bewertet. Nach der AMA-Behandlung waren die Leberbiomarker Aspartataminotransferase (AST), Alaninaminotransferase (ALT) und alkalische Phosphatase (ALP) signifikant erhöht (Abb. 7c, d, f) und das AST/ALT-Verhältnis war deutlich verringert (Abb. 7e).
Dies deutet darauf hin, daß die Leber durch AMA schwer geschädigt wurde. Wichtig ist, daß die ICG-Behandlung die AST-, ALT- und ALP-Spiegel deutlich senkte, was darauf hindeutet, daß ICG den durch AMA verursachten Leberschaden blockieren kann (Abb. 7c–f). Ähnliche Ergebnisse wurden für die Nieren beobachtet, wobei die ICG-Behandlung die renalen Biomarker Blut-Harnstoff-Stickstoff (BUN) und Kreatinin (Cre) bei den mit AMA behandelten Mäusen signifikant reduzierte (Abb. 7g, h).
Die ICG-Behandlung reduzierte auch die Infiltration entzündlicher Zellen und die Nekrose in der Leber von AMA-behandelten Mäusen erheblich (Abb. 7i). Diese Daten wurden quantifiziert und histologische Ergebnisse bestätigten, dass ICG die AMA-induzierte Lebernekrose unterdrücken kann (Abb. 7j), und ähnliche Ergebnisse wurden für mit AMA und ICG behandelte Nieren beobachtet (Abb. 7i, k).
Darüber hinaus führten die Wissenschaftler einen Langzeitüberlebenstest (30 Tage) durch, um zu bewerten, ob ICG auch vor dem durch AMA verursachten Tod schützen kann. Dabei stellten sie fest, daß die ICG-Behandlung das Überleben von mit AMA behandelten Mäusen signifikant verbesserte (Abb. 7l). Darüber hinaus beobachteten sie weder in Kurz- noch in Langzeitstudien offensichtliche Nebenwirkungen bei Mäusen, die nur ICG erhielten, was zeigt, daß diese ICG-Dosis für die Behandlung einer AMA-Vergiftung bei Mäusen sicher ist.
Es wuden auch längere Zeitintervalle (bis zu 12 Stunden) zwischen der AMA-Injektion und der ICG-Behandlung in einem Mausmodell untersucht. Die Ergebnisse zeigten, daß die Zeitintervalle zwischen der AMA-Injektion und der ICG-Verabreichung wichtig für den ICG-Behandlungseffekt waren. Die Intervalle von 8 h und 12 h begrenzten den ICG-Behandlungseffekt, und Intervalle von 1 h und 4 h hatten eine bessere therapeutische Wirksamkeit (ergänzende Abbildung 6). Dies weist darauf hin, daß ICG so früh wie möglich verabreicht werden muss, wenn eine AMA-Vergiftung auftritt.
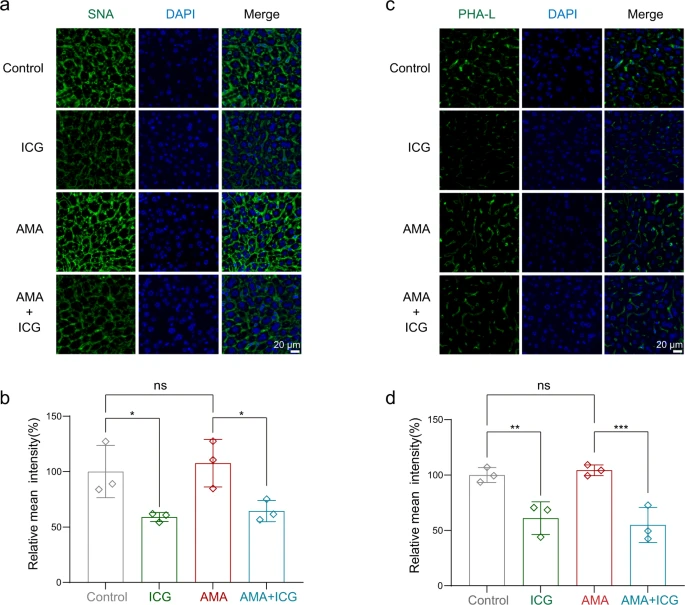
Um die hemmende Wirkung von ICG auf die Proteinglycansynthese bei Tieren zu testen, führte das Team eine Lektinfärbung durch, um die In-vivo-Glykation zu beurteilen. Sie verwendeten Fluorescein-Sambucus-Nigra-Agglutinin (SNA) und Fluorescein-Phaseolus-vulgaris-Leukoagglutinin (PHA-L), um sialylierte Glykane bzw. komplexe Glykane anzufärben, wie bereits berichtet56. Wie erwartet können SNA und PHA-L die glykierten Proteine markieren, die sich auf der Plasmamembran von Leberzellen befinden.
ICG könnte die Fluoreszenzen der SNA- (Abb. 8a, b) und PHA-L-Färbung (Abb. 8c, d) im Vergleich zum Vehikel deutlich reduzieren, was darauf hindeutet, daß ICG die Glykation in vivo effektiv stören könnte. Zusammengenommen zeigen diese Daten, daß ICG ein wirksames Gegenmittel zur Behandlung der AMA-Toxizität bei Mäusen ist.
Diskussion
Unser unvoreingenommenes genomweites CRISPR-Screening hat ergeben, daß sowohl STT3B- als auch die N-Glykan-Biosynthesewege für die AMA-Toxizität erforderlich sind, und diese Daten wurden sowohl genetisch als auch pharmakologisch validiert. Darüber hinaus haben die Forscher durch die Kombination unseres CRISPR-Screenings mit dem In-silico-Screening arzneimittelfähiger Ziele eine von der FDA zugelassene Verbindung ICG entdeckt, die als neuartiges potenzielles Gegenmittel zur Linderung der AMA-Toxizität sowohl in Zellen als auch bei Tieren dienen könnte (Abb. 9). Die gewonnenen Ergebnisse zeigen, daß ein kombinierter Ansatz aus genomweitem CRISPR-Screening in Verbindung mit der In-silico-Arzneimittelvorhersage dabei helfen kann, schnell neue Gegenmittel für medizinisch relevante menschliche Gifte zu identifizieren.
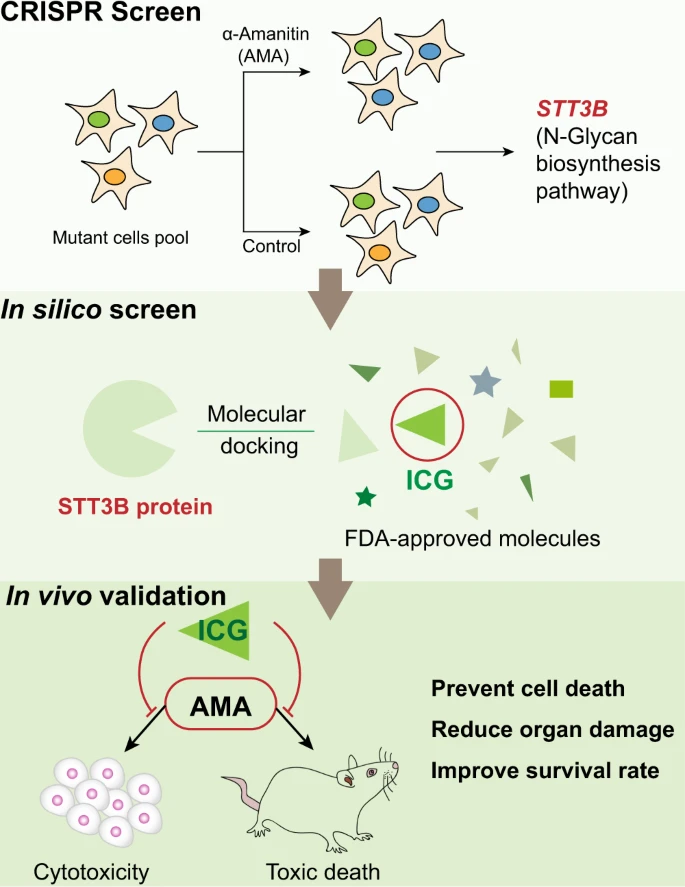
Da der Verzehr von Knollenblätterpilz (Amanita phalloides) zu einer hohen Sterblichkeitsrate führt, besteht ein dringender Bedarf, die molekulare Toxikologie seines wichtigsten toxischen Bestandteils AMA besser zu verstehen. Dies erleichtert somit die Entdeckung wirksamer Gegenmittel zur Behandlung von Pilzvergiftungen.
Mithilfe eines genomweiten CRISPR-Cas9-Screenings haben wir mehrere wichtige Gene und Signalwege identifiziert, die am AMA-induzierten Zelltod beteiligt sind. Um die Fähigkeit des Screenings zur Identifizierung von Schlüsselfaktoren zu unterstützen, gehörten einige der bekannten Mechanismen, die an der AMA-Toxizität beteiligt sind, wie Apoptose und RNA-Polymerase-II-Hemmung, zu den Signalwegen und GO-Begriffen. Einer unserer wichtigsten Wege, die N-Glykan-Biosynthese, wurde sowohl pharmakologisch als auch genetisch validiert. Die N-verknüpfte Glykosylierung spielt eine entscheidende Rolle bei der Proteinfaltung und dem Proteintransport, die an einer Vielzahl biologischer Erkennungsereignisse beteiligt sind34.
Es wurde berichtet, daß bakterielle Toxine37,57 und Viren23,58 N-Glykane binden und diese Wechselwirkung den Eintritt in Zielzellen erleichtert. In diesem Fall stellten wir die Hypothese auf, dass die Blockierung von N-Glykanen von AMA-Transportern die Erkennung von AMA behindern könnte, was zur Resistenz beiträgt. STT3B ist ein Schlüsselenzym für die N-Glykan-Biosynthese und gilt als therapeutisches Ziel für die Behandlung von Krebserkrankungen42. Es wurde jedoch keine von der FDA zugelassene Verbindung zur Hemmung der N-Glykan-Biosynthese oder von STT3B identifiziert. Durch In-silico-Screening von FDA-zugelassenen Molekülen konnten die Forscher erfolgreich nachweisen, daß ICG ein potenzieller STT3B-Inhibitor zur Hemmung der N-Glykan-Biosynthese ist. ICG ist ein wasserlöslicher Nahinfrarot-Fluoreszenzfarbstoff, der seit Jahrzehnten häufig als Diagnosemittel zur Messung der Leberfunktion, des Herzzeitvolumens und der Augenangiographie beim Menschen eingesetzt wird59,60.
Es hat sich herausgestellt, daß ICG den AMA-induzierten Zelltod wirksam verhindern und die Resistenz gegen AMA in vitro erhöhen kann. Der enterohepatische Zyklus von AMA könnte den klinischen Verlauf einer Amatoxinvergiftung bei Menschen und Tieren erheblich beeinflussen61. Die Unterbrechung des enterohepatischen Kreislaufs wie die Gallendrainage ist zu einer alternativen Entgiftungsmethode geworden62. Da ICG den Eintritt von AMA verhindern kann, kann ICG die AMA-Rezirkulation blockieren und die AMA-Toxizität verhindern, was teilweise die therapeutische Wirkung von ICG erklärt.
HepG2-Zellen wurden zur Untersuchung der Hepatotoxizität63 und der molekularen Mechanismen der AMA-Zytotoxizität7 verwendet. HepG2-Zellen zeigen eine relativ geringe Expression von OATP1B364 und exprimieren kein NTCP65, während diese beiden Proteine als Haupttransporter für die AMA-Aufnahme gelten. Daher weisen HepG2-Zellen im Vergleich zu anderen gängigen Laborzelllinien oder primären Hepatozyten eine geringere Empfindlichkeit gegenüber AMA-Zytotoxizität auf66. Dennoch können in unserer Studie HepG2-Zellen immer noch durch AMA abgetötet und durch ICG gerettet werden, was darauf hindeutet, daß es unbekannte Transporter gibt, die für den AMA-Transport in Zellen verantwortlich sind.
Interessanterweise wurde ICG auch durch OATP1B3 und NTCP67 in Zellen transportiert. Unsere Ergebnisse schließen die Möglichkeit aus, dass ICG hauptsächlich durch kompetitive Bindung mit OATP1B3 und NTCP wirkt, um die Toxizität von AMA zu blockieren. Darüber hinaus zeigte unsere Studie, dass ICG ähnliche Schutzwirkungen in AMA-behandelten primären Hepatozyten-Organoiden hat. In dieser Studie haben die Wissenschaftler sich auf das 4-Stunden-Intervall zwischen der AMA-Injektion und der ICG-Behandlung konzentriert, da dieses 4-Stunden-Intervall das gleiche war wie das, das in früheren Studien verwendet wurde, um tatsächliche Vergiftungs- und Behandlungsszenarien beim Menschen nachzuahmen5,52,68.
Dies wird es zukünftig erleichtern, die gewonnenen Ergebnisse mit denen anderer hinsichtlich der Wirksamkeit von ICG bei der Behandlung von AMA-Toxizität zu vergleichen. ICG hat ein großes Potenzial für die Behandlung von AMA-Vergiftungen bei Mäusen gezeigt und die ICG-Behandlung kann AMA-induzierte Schäden in Leber und Niere, den beiden wichtigsten AMA-Zielorganen, erheblich abschwächen, was zu einer Verbesserung des Überlebens führt. Darüber hinaus wurde beobachtet, daß ICG seine Behandlungswirkung auf die AMA-Toxizität verliert, wenn es 8 und 12 Stunden nach der AMA-Injektion verabreicht wird.
Dies kann daran liegen, daß AMA in den ersten Stunden der Zytotoxizität irreversible Schäden verursacht hat, die durch eine ICG-Behandlung nicht behoben werden können. Dies legt nahe, daß ICG so früh wie möglich während der Behandlung verabreicht werden sollte. Insgesamt zeigte sich, daß durch die Kopplung der funktionellen genomischen Charakterisierung des gesamten Genoms mit der In-silico-Arzneimittelvorhersage schnell medizinisch relevante Prozesse definieren lassen und dann gezielt darauf abzielen können.
Ethische Aussage
Alle Tierversuche wurden vom Institutional Animal Care and Use Committee (IACUS) der Sun Yat-Sen-Universität genehmigt (Genehmigungsnummer: SYSU-IACUC-2022-000469).
Zelllinien und Zellkultur
HAP1-Zellen wurden von Horizon Discovery erhalten. HEK293T- und HepG2-Zelllinien wurden von der American Type Culture Collection (ATCC) erhalten. Alle Zelllinien wurden routinemäßig mit dem Mycoplasma Stain Assay Kit (#C0296, Beyotime) mykoplasmenfrei getestet und durch Short Tandem Repeat (STR)-Profiling authentifiziert. HAP1-Zellen wurden in Iscoves modifiziertem Dulbecco-Medium (IMDM; Gibco), ergänzt mit 10 % fötalem Rinderserum (FBS; NEWZERUM) und 1 % Penicillin-Streptomycin (Hyclone), kultiviert. HEK293T- und HepG2-Zellen wurden in Dulbeccos modifiziertem Eagle-Medium (DMEM, Gibco), ergänzt mit 10 % FBS und 1 % Penicillin-Streptomycin, kultiviert.
Zelllebensfähigkeitstest
Trypsinisierte Zellen (1,5 × 104) wurden in jede Vertiefung einer Platte mit 96 Vertiefungen ausgesät. Nach 24 Stunden wurden verschiedene Konzentrationen der Verbindungen hinzugefügt und die Zellen wurden weitere 48 oder 72 Stunden lang inkubiert. KIF (#K919109) und NGI-1 (#N873007) wurden von Macklin erhalten. Nach der Inkubation wurde das Medium aus jeder Vertiefung abgesaugt und 100 μl frisches Medium, das ein 10 %iges Cell Counting Kit-8 (CCK8, #K1018, APExBIO) enthielt, in die Vertiefungen gegeben und 2 Stunden lang bei 37 °C inkubiert.
Die Absorption wurde bei 450 nm mit einem Mikroplatten-Spektrophotometer (BioTek) gemessen. Die Zellüberlebensdaten jeder medikamentös behandelten Gruppe wurden auf die Vehikelgruppe normalisiert und die Überlebensdaten als „relative Zelllebensfähigkeit“ ausgedrückt. Die Lebensfähigkeit von Calcein/PI-Zellen wurde gemäß dem Protokoll des Herstellers (#C2015M, Beyotime) bewertet und unter Fluoreszenzmikroskopie (Nikon) beobachtet.
Lentivirus-Produktion
Um Lentivirus zu erzeugen, wurde die Brunello-Bibliothek (#73179, Addgene) mit den Verpackungsplasmiden pMD2.G (#12259, Addgene) und psPAX2 (#12260, Addgene) co-transfiziert. Kurz gesagt wurde ein T75-Kolben mit 80 % konfluenten HEK293T-Zellen in Opti-MEM (#31985070, Gibco) unter Verwendung von 8,8 μg der lentiCRISPRv2-Plasmidbibliothek, 4,4 μg pMD2.G, 6,7 μg psPAX2 und 32 μL Lipo8000TM (#C0) transfiziert 533 , Beyotime). Die Zellen wurden 8 Stunden lang inkubiert und dann wurde das Medium durch DMEM mit 10 % FBS und 1 % Penicillin-Streptomycin ersetzt. Das Virus wurde 48 Stunden nach der Transfektion geerntet, das Medium wurde aufgefüllt und eine zweite Ernte erfolgte 72 Stunden nach der Transfektion. Virusüberstände wurden gesammelt und durch einen 0,45-μm-Filter mit extrem geringer Proteinbindung (Merck Millipore) filtriert. Aliquote wurden bei –80 °C gelagert.
Zelltransduktion mithilfe der Brunello-Bibliothek
Infektionen wurden in einer 12-Well-Platte mit 2,0 × 106 Zellen pro Well durchgeführt. In jede Vertiefung wurden unterschiedliche Virenmengen gegeben. Nach 24 Stunden wurden die Zellen trypsiniert und jede Vertiefung in zwei Vertiefungen aufgeteilt. Und ein Replikat wurde 3 Tage lang mit 1 μg/ml Puromycin (#P8230, Solarbio) behandelt, bis unter Bedingungen ohne Virus keine lebensfähigen Zellen mehr vorhanden waren. Abschließend wurde die Lebensfähigkeit der Zellen für jede Bedingung mithilfe eines CCK8-Assays quantifiziert. Das Virusvolumen mit einem MOI von etwa 0,3 wurde für das nächste groß angelegte Screening verwendet.
HAP1-Zellen-AMA-Resistenztest
Um die Abdeckung von >500 Zellen sicherzustellen, die jeweils 77.441 sgRNA mit einer MOI ≈ 0,3 exprimieren, wurden 1,3 × 108 HAP1-Zellen wie oben beschrieben unter Verwendung von 12-Well-Platten mit 2 × 106 Zellen pro Well transduziert. Puromycin wurde den Zellen 24 Stunden nach der Transduktion zugesetzt und 7 Tage lang beibehalten. Die Zellen wurden nach 3-tägiger Inkubation mit Puromycin in größeren Flaschen zusammengefasst. Am 7. Tag wurden die Zellen in zweifacher Ausführung in die Behandlungsbedingungen aufgeteilt, mit einem Minimum von 4 × 107 Zellen pro Wiederholung. Zwei Replikate wurden in einem Komplettmedium mit 1,5 μM AMA (#A4548, APExBIO) kultiviert, und zwei weitere Replikate wurden in einem regulären Komplettmedium kultiviert. Es wurden entweder Replikate durchgeführt oder alle 2–3 Tage wurde frisches Medium hinzugefügt. Die mutierten Zellpools wurden 7 Tage lang mit AMA behandelt und die überlebenden Zellen wurden gewonnen und für die genomische DNA-Analyse geerntet.
Genomische DNA-Sequenzierung
Genomische DNA (gDNA) wurde mit TIANamp Genomic DNA Kits (#DP304, TIANGEN) gemäß dem Protokoll des Herstellers isoliert. Die sgRNA-Sequenzen wurden mit High-Fidelity 2X PCR Master Mix (#M0541L, NEB) amplifiziert. PCR-Produkte wurden mit Illumina (PE150) von Novogene Technology (Peking, China) gelextrahiert, quantifiziert, gemischt und sequenziert. Die Anreicherung von sgRNAs und Genen wurde mit MAGeCK (Version 0.5.9.2)69 analysiert, indem die Lesezahlen von Zellen nach der AMA-Selektion mit den Zahlen aus passenden, nicht ausgewählten Zellpopulationen verglichen wurden.
Genontologie (GO) und Signalweganreicherungsanalyse
GO-Begriffe im Bildschirm wurden mit DAVID (https://david.ncifcrf.gov/summary.jsp) analysiert. KEGG-Pfade wurden mit Webgestalt (http://www.webgestalt.org/) analysiert. Anreicherungsnetzwerkpfade wurden mithilfe von String (https://cn.string-db.org/) und Cytoscape (https://cytoscape.org/) generiert.
Etablierung von Knockout-Zelllinien
sgRNAs aus der Elternbibliothek wurden in pLentiCRISPRv2 (# 52961, Addgene) kloniert. Die Kontroll-sgRNA wurde aus der Elternbibliothek verwendet (Ergänzungstabelle 1). Lentiviren wurden wie oben beschrieben hergestellt und transduzierte HAP1- oder HepG2-Zellen wurden 24 Stunden nach der Infektion mit Puromycin selektiert. Nach 7 Tagen wurde Puromycin entfernt und die Zellen konnten sich vor der Analyse drei weitere Tage lang erholen.
RNA-Extraktion und quantitative Echtzeit-PCR-Analyse
Die Gesamt-RNA wurde mit einem RNA Quick Purification Kit (#RN001, YIBIN) extrahiert. Die cDNA wurde aus 500 ng Gesamt-RNA mit PrimeScript RT Master Mix (#RR037A, Takara) gemäß den Anweisungen des Herstellers synthetisiert. Quantitative PCR (qPCR) wurde mit TB Green (#RR820A, Takara) gemäß dem Protokoll des Herstellers mit LightCycler96 (Roche) durchgeführt.
Hochleistungsflüssigkeitschromatographie (HPLC)
Die HPLC-Analyse wurde mit SHIMADZU LC-20AT und einer 250 mm × 4,6 mm Flüssigkeitschromatographiesäule (5 μm, Phenomenex) durchgeführt. Ammoniumacetat (50 mM Essigsäure, pH 5,5), Acetonitril und Methanol (80/10/10; v/v/v) wurden nach einer früheren Studie als mobile Phase verwendet70. Eine isokratische Elution wurde mit einer Flussrate von 1,0 ml/min und einer Säulentemperatur von 35 °C durchgeführt. Insgesamt wurden 2 × 107 Zellen, die 8 h lang mit AMA behandelt wurden, gesammelt und dann durch Ultraschallbehandlung bei 200 W (3 s Arbeit/3 s Pause) für 1 min auf Eiswasser aufgebrochen. Diese Mischung wurde 30 Minuten lang bei 15.000 g zentrifugiert. Der Überstand wurde gesammelt und durch einen 0,22-μm-Filter (Merck Millipore) filtriert. Insgesamt wurden 10 μL Probe in das HPLC-System injiziert. Für quantitative Analysen wurden Chromatogramme bei 303 nm integriert. Die Peakfläche wurde mit der LabSolutions-Software (Version 1.26) analysiert.
Western-Blot-Analyse
Die Zellen wurden in einem Lysepuffer (#FD009, Fdbio) geerntet, der einen Proteaseinhibitor-Cocktail (Roche) enthielt. Die gesamten Zelllysate wurden 10 Minuten lang bei 4 °C und 15.000 g zentrifugiert, um Zelltrümmer zu entfernen. Die Proteinkonzentrationen wurden mit dem BCA Protein Assay (#P0010, Beyotime) bestimmt. Die Proteinüberstände wurden mit 5×Ladepuffer (FD006, Fdbio) gemischt und 10 Minuten lang auf 100 °C erhitzt. Die Proteine (20 μg) wurden auf 10 % SDS-Polyacrylamidgelen elektrophoretisch aufgetrennt, auf PVDF-Membranen übertragen und über Nacht bei 4 °C mit spezifischen Primärantikörpern inkubiert. Nach dem Waschen wurden die Membranen 1 Stunde lang mit Meerrettichperoxidase (HRP)-konjugierten Sekundärantikörpern inkubiert. Immunoblots wurden mit dem ChemiDocTM-Bildgebungssystem (Bio-Rad, Version 2.4.0.03) unter Verwendung des verbesserten Chemilumineszenzsubstrats (FD8000, Fdbio) sichtbar gemacht.
Für den Proteinnachweis wurden die folgenden Antikörper verwendet: monoklonaler Maus-Antikörper, der OATP1B3 erkennt (#66381-1-Ig, 1:5000, Klon-Nr. 1D9A4), monoklonaler Maus-Antikörper, der GRP78 erkennt (#66574-1-Ig, 1:5000, Klon-Nr .1D6F7), polyklonaler Kaninchen-Antikörper, der IRE1 erkennt (#27528-1-AP, 1:1000), monoklonaler Maus-Antikörper, der GM130 erkennt (#66662-1-Ig, 1:5000, CloneNo.2A4F11), polyklonaler Kaninchen-Antikörper, der ARF4 erkennt( #11673-1-AP, 1:1000) wurden von Proteintech erworben. Der polyklonale Kaninchen-Antikörper, der NTCP erkennt (#ABP53103, 1:1000), wurde von Abbkine erworben. Der Mausantikörper, der β-Tubulin erkennt (#FD0064, 1:5000), wurde von Fdbio erworben. Ziegen-Anti-Maus-IgG(H + L)-HRP (#BS12478, 1:5000) und Ziegen-Anti-Kaninchen-IgG(H + L)-HRP (#BS13278, 1:5000) wurden von Bioworld gekauft.
In-silico-Screening von FDA-zugelassenen Medikamenten
Die beiden Bibliotheken von FDA-zugelassenen Verbindungen wurden von ZINC (1615 Liganden) bzw. Drugbank (1586 Liganden) heruntergeladen. Die chemische Datenbank wurde mithilfe des MMFF94-Kraftfelds (steilster Abstieg) im Open-Source-Softwarepaket OpenBabel (http://openbabel.org/) energieminimiert und im mol2-Format gespeichert. Die Verbindungsbibliothek wird dann vorverarbeitet und im pdbqt-Format mit Prepare_ligand4.py aus den AutoDock-Tools (https://ccsb.scripps.edu/mgltools/) gespeichert.
Die Kristallstruktur von STT3B wurde aus RCSB PDB (PDB-ID: 6S7T)71 erhalten. Das STT3B wurde von PyMol (http://pymol.org/2/) vorverarbeitet, um Wasser und Liganden zu entfernen, und von AutoDock-Tools, um polare Wasserstoffe und Kollman-Ladung hinzuzufügen. Nach diesen Vorgängen wurde STT3B dann im pdbqt-Format gespeichert. Im STT3B gibt es zwei Bindungstaschen. Das Rasterfeld der Bindungsstelle wurde mithilfe der Rastereinstellungsfunktion der AutoDock-Tools visuell definiert. Die Gitterbox aus zwei Bindungstaschen wurde durch gebundenes Peptid und gebundenes Dolichylphosphat in 6S7T definiert. Für die STT3B-Tasche 1 betrugen die Gittergrößenabmessungen 23,25 × 22,5 × 24, wobei der Punkt (173,747, 153,363, 152,254) als Mittelpunktskoordinaten festgelegt wurde; Für die STT3B-Tasche 2 betrugen die Gittergrößenabmessungen 23,25 × 24,75 × 24,75, wobei der Punkt (159,012, 143,831, 166,532) als Mittelpunktkoordinaten festgelegt wurde.
Der Andockvorgang wurde mit Smina (einem Zweig von AutoDock Vina, https://vina.scripps.edu/)72 durchgeführt. Molekulare Docking-Parameter werden wie folgt verwendet: Vollständigkeit = 8, num_modes = 10, Energiebereich = 3, min_rmsd_filter = 1. Es wurde ein schrittweises Screening von FDA-zugelassenen Arzneimitteln in den ZINC- und Drugbank-Datenbanken durchgeführt. Zunächst wurden die Verbindungen jeweils an die beiden Bindungstaschen von STT3B angedockt. Dann wurden die Verbindungen nach ihrer minimierten Affinität eingestuft. Es wurden die 100 besten Liganden mit minimierter Affinität erhalten. Anschließend wurde der vorhergesagte IC50-Wert der Verbindungen mit der auf einem neuronalen Netzwerk basierenden Bewertungsfunktion (NNScore2)73 berechnet. Am Ende wurden die 34 besten Verbindungen für die In-vitro-Validierung ausgewählt. Aufgrund der Nichtverfügbarkeit einiger Verbindungen wurden nur 24 Verbindungen auf ihren zellulären Schutz gegen AMA-Toxizität getestet.
Organoidkultur
Die Leberorganoide der Mäuse wurden gemäß zuvor beschriebenen Protokollen mit einigen Modifikationen erzeugt74,75. Kurz gesagt, die Lebern von CD-1-Mäusen (20–30 g) wurden in Würfel von 1–2 mm3 zerlegt und zweimal in PBS gewaschen. Die Gewebefragmente wurden in Verdauungspuffer (1 mg/ml Kollagenase I, 0,1 mg/ml Hyaluronidase, 0,1 mg/ml DNase I) 1,5 h lang bei 37 °C inkubiert. Nach der Verdauung wurde die Gewebesuspension mit einem 40-μm-Zellsieb filtriert, anschließend zentrifugiert und mit PBS resuspendiert. Einzelzellsuspensionen wurden zunächst mit hoher Dichte ausgesät und nach etwa einer Woche in vollständigem Organoidmedium (DMEM/F12 mit 5 µg/ml Insulin, 250 µg/ml Amphotericin B, 10 µg/ml Gentamicin, 0,125 µg) mit niedrigerer Dichte erneut ausgesät /ml EGF, 25 ng/ml Hydrocortison und 10 μm Y-27632).
Intrazelluläre Lokalisierungsanalyse
HAP1- und HepG2-Zellen wurden bis zu einer Konfluenz von ~50 % in die Kulturschale ausplattiert und 12 Stunden lang mit ICG (#H20055881, Dandong Pharmaceutical Company) inkubiert. Die Zellen wurden dreimal mit PBS gewaschen und ER wurde gemäß dem Protokoll des Herstellers (#C1042S, Beyotime) mit ER-Tracker-Grün markiert, und der Zellkern wurde mit 4′,6-Diamidino-2-phenylindol (DAPI) blau gefärbt. Fluoreszenzbilder wurden durch konfokale Laser-Scanning-Mikroskopie erhalten.
Luciferase-Reporter-Assay
Die ER-LucT-Sequenz und die LucT-Sequenz aus einer früheren Studie49 wurden von Kidan Bio Co. Ltd. synthetisiert und in pcDNA3.1(+) kloniert. Plasmidtransfektionen wurden mit Lipo8000TM durchgeführt. Nach 48 Stunden wurde ICG zu den transfizierten HAP1- und HepG2-Zellen gegeben. Luciferase-Aktivitätstests wurden mit dem Firefly Luciferase Reporter Gene Assay Kit gemäß dem Protokoll des Herstellers (#RG005, Beyotime) mit dem Lumineszenz Quick Read (Promega) durchgeführt.
Mäuse
Alle Mäuse waren männliche CD-1-Mäuse mit einem Gewicht von 20–30 g (4–5 Wochen alt) und wurden vom Laboratory Animal Center der Sun Yat-Sen-Universität gekauft. Die Mäuse wurden in einer speziellen pathogenfreien Einrichtung mit kontrollierter Temperatur (23 ± 2 °C), Luftfeuchtigkeit (50–65 %) und einem Hell-Dunkel-Zyklus von 12 Std./12 Std. gehalten. Mäuse hatten freien Zugang zu Futter und Wasser.
Körpergewicht, motorische Aktivität, Atemnot und das allgemeine Wohlbefinden der Tiere wurden täglich beobachtet. Die Mäuse wurden gemäß den von der IACUC zugelassenen Anästhesiemethoden mit 1 % Pentobarbital-Natrium und anschließender Zervixluxation eingeschläfert.
Kurzzeitstudie an Mäusen (24 h)
Die Mäuse wurden zufällig in 4 Gruppen verteilt (n = 6) und wie folgt behandelt: (i) Kontrollgruppe (0,9 % NaCl, i.p. bei 0, 4, 8 und 12 Stunden); (ii) ICG-Gruppe (0,9 % NaCl, i.p. bei 0 h; 5 mg/kg ICG, i.v. bei 4, 8 und 12 h); (ii) AMA-Gruppe (0,33 mg/kg AMA i.p. bei 0 h; 0,9 % NaCl, i.v. bei 4, 8 und 12 h); (iii) AMA + ICG-Gruppe, (0,33 mg/kg AMA i.p. bei 0 h; 5 mg/kg ICG i.p. bei 4, 8 und 12 h).
Langzeitstudie an Mäusen (30 Tage)
Die Mäuse wurden zufällig in 4 Gruppen eingeteilt (n = 6) und demselben Verabreichungsprotokoll wie in der Kurzzeitstudie unterzogen. Körpergewicht, motorische Aktivität, Atemnot und allgemeines Wohlbefinden der Tiere wurden 30 Tage lang täglich beobachtet.
In-vivo-Fluoreszenzbildgebung
Insgesamt wurden 5 mg/kg ICG intravenös injiziert und Bilder wurden 0, 10, 20, 40, 60 und 120 Minuten nach der Injektion mit einem In-vivo-Bildgebungssystem (PerkinElmer) aufgenommen.
Blutbiomarker
24 Stunden nach der AMA-Injektion wurden alle Tiere betäubt und eingeschläfert. Das Blut wurde in EDTA-haltige Röhrchen entnommen und sofort 10 Minuten lang bei 3000 g (4 °C) zentrifugiert. Der Plasmaüberstand wurde in Röhrchen gesammelt und bis zur Bestimmung bei –80 °C gelagert. AST, ALT, ALP, BUN und Cre wurden vom Guangdong Engineering & Technology Research Center for Disease-Model Animals der Sun Yat-sen University gemessen.
Histologische Analyse von Leber und Niere
Nach der Blutentnahme wurden Leber und Nieren entnommen und gewogen. Leber- und Nierensegmente wurden zur H&E-Färbung durch Servicebio-Technologie in 4 % Paraformaldehyd gelegt. Der Grad der Entzündung und der Nekrose der Objektträger wurde blind anhand des folgenden Kriteriums halbquantifiziert: Grad 0 = keine Veränderung gegenüber dem Normalwert; Grad 1 = sehr mild (die Veränderungen liegen knapp außerhalb des normalen Bereichs); Grad 2 = leicht (Läsionen können beobachtet werden, sind aber nicht schwerwiegend); Grad 3 = mittel (Die Läsionen sind offensichtlich und wahrscheinlich schwerwiegender) und Grad 4 = schwer (die Läsion hat das gesamte Gewebe und Organ besetzt).
Lektinfärbung
Leberschnitte aus verschiedenen Mäusegruppen wurden geschnitten, entparaffiniert und 30 Minuten lang bei Raumtemperatur mit verdünnten Lektinen gefärbt. Fluorescein-SNA (bindet Sialinsäure, FL-1301-2, 1:200) und Fluorescein-PHA-L (bindet komplexe Glykane, #FL-1111-2, 1:200) wurden von Vector Laboratories erworben. Die Objektträger wurden dann mit DAPI gefärbt und zweimal in PBST (phosphatgepufferte Kochsalzlösung mit Triton X-100) gewaschen und mittels Fluoreszenzmikroskopie (Nikon) abgebildet.
Statistik und Reproduzierbarkeit
Zur Vorbestimmung der Stichprobengröße wurde keine statistische Methode verwendet. Es wurden keine Daten von den Analysen ausgeschlossen. Die Daten werden als Mittelwert ± Standardabweichung (S.D.) dargestellt. Statistische Analysen wurden mit der GraphPad Prism 9-Software (Version 9.0.0) durchgeführt. p < 0,05 wurde als statistisch signifikant angesehen. *p < 0,05; **p < 0,01; ***p < 0,001; ****p < 0,0001; ns, nicht signifikant. In den Legenden wurden biologische Replikate und die Anzahl unabhängiger Experimente angegeben. Alle Experimente, die als repräsentative mikroskopische Aufnahmen oder Gele präsentiert wurden, wurden mindestens dreimal mit ähnlichen Ergebnissen wiederholt.
Zusammenfassung der Berichterstattung
Weitere Informationen zum Forschungsdesign finden Sie in der mit diesem Artikel verlinkten Nature Portfolio Reporting Summary.
Datenverfügbarkeit
Die in dieser Studie generierten DNA-Sequenzierungsdaten wurden in der Gene Expression Omnibus (GEO)-Datenbank unter dem Zugangscode GSE226447 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226447) hinterlegt. . ZINC (https://zinc20.docking.org/) und Drugbank (https://go.drugbank.com/) sind öffentlich verfügbare Datensätze. Die Daten, die die Ergebnisse dieser Studie stützen, sind im Artikel und in der Datei mit ergänzenden Informationen verfügbar. Quelldaten werden in diesem Dokument bereitgestellt. Quelldaten werden mit diesem Dokument bereitgestellt.